Using genetically modified organisms is a staple in today's research laboratories throughout the life sciences. For this research, a 'wild type' reference strain is indispensable to compare the manipulated organisms to. Therefore, every laboratory keeps such strains, which are then referenced in the literature by their common name: C57BL/6 is such a strain in mice, N2 in the nematode C. elegans or Canton S in the fruit fly Drosophila.
However, each strain is separate from each other such strain as these reference strains are rarely ever exchanged or mixed. In fact, as copies are drawn from a laboratory's reference strain, new genetic subsamples of each particular variant are constantly created anew. In this way, such sub-strains have proliferated with the increase in the number of researchers world wide. In several organisms, the common name for these strains has proven misleading: every laboratory may hold a different strain, while there is little acknowledgment of their difference in the literature.
Online fly tracking and open data: Buridan's Paradigm
During routine behavioral work with Canton S fruit fly strains from different laboratories, we noticed striking differences in the behavior of these strains. The experiment we used in Berlin was a simple experiment to study locomotor behavior in flies, Buridan's paradigm. This short video clip explains the basic idea behind the experiment:
Striking differences
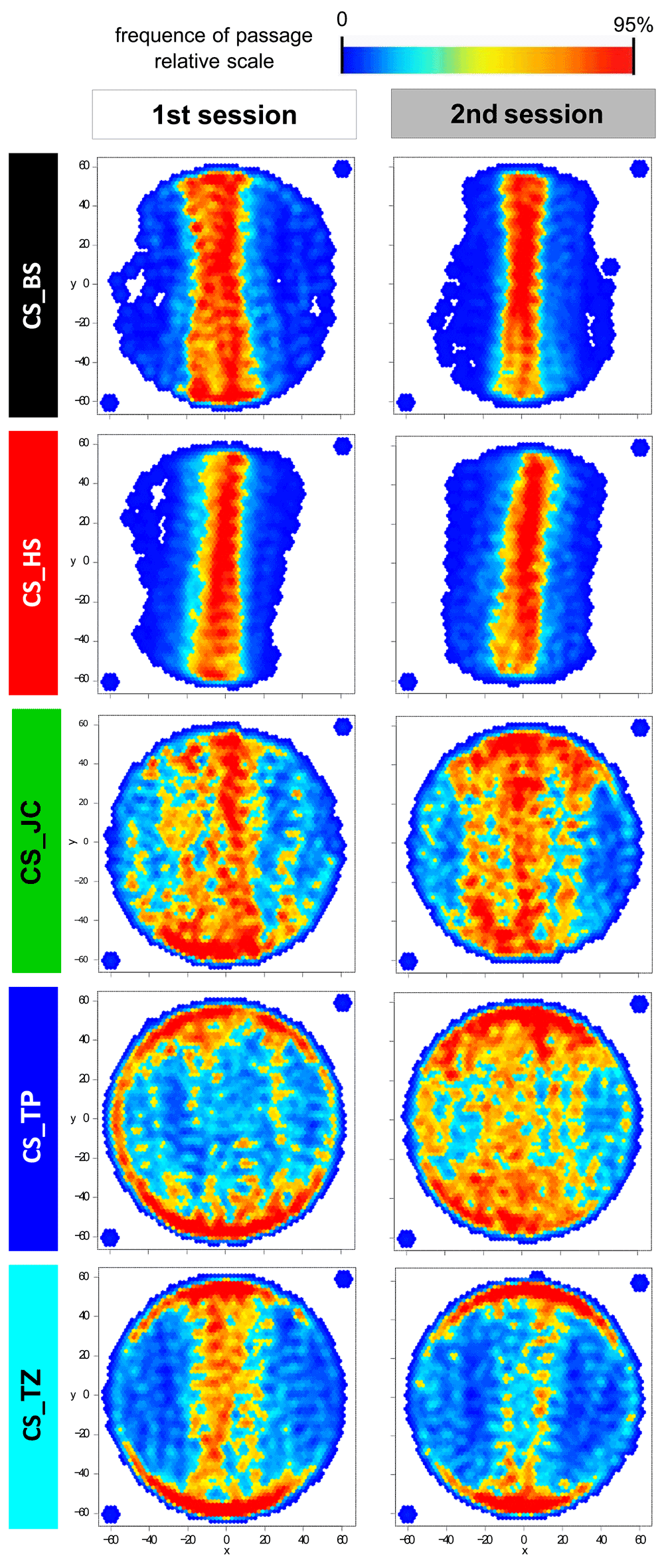
What we found was that the different sub-strains differed with respect to their spatial behavior towards the two stripes. Some strains walked between the stripes with robotic stereotypy, while other seemed to hardly even notice them, and other strains in-between. The transition plots below show how the different sub-strains (rows) walked in two different experiments, spaced one year apart (columns). The plots are arranged such that the stripes are located North and South in all plots (click for larger version):
Open Science
After publishing these findings, we were contacted by Casey Bergman, with the suggestion to collaborate on studying the genomes of our five strains. I enthusiastically agreed and we started a fully open project, where all methods, data and discussion was taking place on GitHub. I started by testing 20 females of each strain in Buridan's paradigm before extracting their DNA and sending it to Casey. His sequencing very quickly revealed that each strain could be distinguished from all other strains already by just clicking through the genomes in a very simple genome browser.
Before we could perform any additional analysis, we were contacted by Nelson Lau who had done a thorough analysis of all the approximately 30k transposable elements (aka. jumping genes) in these strains. His analysis confirmed what seemed so apparent to the naked eye: there are substantial differences in the genomes between each of these strains.
The reason Nelson contacted us was to propose to make us authors of his new publication, even though he had just used our publicly posted data. I protested, emphasizing that I had just extracted the DNA. I argued that I be at most placed inthe acknowledgment section for "technical assistance". We argued back and forth for quite some time, until I finally gave up when his argument was that he wanted to use ouzr names on the author list to demonstrate that publicly depositing raw data can be a rewarding practice that more colleagues ought to follow. Of course we all read and commented on the manuscript, so now my contribution is somewhat more intellectual than just extracting the DNA, but I still feel sort of uneasy for this, in my opinion still ill-deserved place on a manuscript which was not mine. I also still think I should not get rewarded for a practice that should be a matter of course.
Be that as it may, I am very excited about what we have learned from these projects and the potential new insights the genomes and the behavioral data may yet provide. For everyone interested in learning more about this research, we have three summaries here, here and here, besides the two publications, of course:
Abstract: We collected five sub-strains of the standard laboratory wild-type Drosophila melanogaster Canton Special (CS) and analyzed their walking behavior in Buridan's paradigm using the CeTrAn software. According to twelve different aspects of their behavior, the sub-strains fit into three groups. The group separation appeared not to be correlated with the origin of the stocks. We conclude that founder effects but not laboratory selection likely influenced the gene pool of the sub-strains. The flies’ stripe fixation was the parameter that varied most. Our results suggest that differences in the genome of laboratory stocks can render comparisons between nominally identical wild-type stocks meaningless. A single source for control strains may settle this problem.
Abstract: To understand how transposon landscapes (TLs) vary across animal genomes, we describe a new method called the Transposon Insertion and Depletion AnaLyzer (TIDAL) and a database of >300 TLs in Drosophila melanogaster (TIDAL-Fly). Our analysis reveals pervasive TL diversity across cell lines and fly strains, even for identically named sub-strains from different laboratories such as the ISO1 strain used for the reference genome sequence. On average, >500 novel insertions exist in every lab strain, inbred strains of the Drosophila Genetic Reference Panel (DGRP), and fly isolates in the Drosophila Genome Nexus (DGN). A minority (<25%) of transposon families comprise the majority (>70%) of TL diversity across fly strains. A sharp contrast between insertion and depletion patterns indicates that many transposons are unique to the ISO1 reference genome sequence. Although TL diversity from fly strains reaches asymptotic limits with increasing sequencing depth, rampant TL diversity causes unsaturated detection of TLs in pools of flies. Finally, we show novel transposon insertions negatively correlate with Piwi-interacting RNA (piRNA) levels for most transposon families, except for the highly-abundant roo retrotransposon. Our study provides a useful resource for Drosophila geneticists to understand how transposons create extensive genomic diversity in fly cell lines and strains.